Phase Equilibria at High Pressure

Outline
High Pressure Phase Equilibrium Studies
Phase Equilibria Relevant to the Deep Earth
Introduction
Studies of phase equilibria are highly relevant to many areas of geosciences because in most cases, mineral systems have the time to reach thermodynamic equilibrium and lack the energy input needed to sustain disequlibrium. With increasing pressure, most crystal structures that are stable at low pressure (e.g. 1 bar) are replaced by denser structures. This has important implications for Earth's interior as well as for the synthesis of novel materials for industrial applications. A significant area of mineral physics research is therefore dedicated towards discovering new phases that may be present either in the deep earth or within other planets and characterizing their physical and chemical properties. Equilibrium relations between phases at high pressure are governed by the same thermodynamic principles that operate at low pressure. There are many good books and resources for brushing up on these concepts including those found elsewhere in the SERC website (see links below). Concepts that are particularly important to understand are: Gibbs Free energy, enthalpy, entropy, chemical activity, Clausius Clapeyron equation, endothermic and exothermic reactions and the phase rule. If one is going to delve into multicomponent systems it is also a good idea to be familiar with Schreinemakers method.High Pressure Phase Equilibrium Studies
There are a number of different types of high pressure studies related to phase equilibria. There are true phase equilibrium studies that seek to characterize the range of pressure, temperature or chemical activity (e.g. for O2) over which a given phase (or phase assemblage) is stable. Synthesis studies seek to discover and produce new phases. However, they are distinct from phase equilibrium studies. There are a number of circumstances that can lead to minerals forming outside their thermodynamic stability field. Another major area of interest is phase transformation mechanisms and kinetics. Reversals: the difference between synthesis and phase equilibrium studies
One of the most important features of a good phase boundary determination is the reversal. A reversal is a pair of experiments where the reactants and products in one experiment become the products and reactants in another experiment. The position of the phase boundary can be said to be somewhere between these bracketing experiments. In-situ experiments (e.g. synchrotron x-ray diffraction) are particularly efficient for conducting phase equilibrium studies because the transformation from one phase assemblage to the other can be run first one way and then back the other at a number of pressure and temperatures during a single experiment.
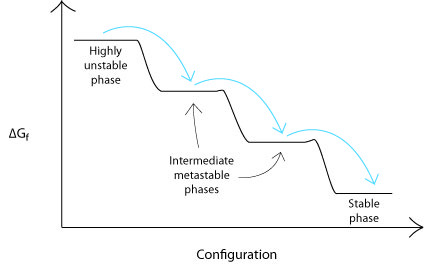 Ostwald's Step Rule
Ostwald's Step Rule
Experimental Concerns
The quality of a phase equilibrium study is highly dependent on how well known P, T and chemical activity are in the experiment. Discussions of uncertainty in P and T can be found in the multi-anvil and diamond anvil pages. Knowing the chemical activity of most elements in an experiment is quite straightforward; if the element was present in the capsule both before and after the experiment its activity is be assumed to be one.


Discovery and Synthesis of New Phases

Phase Equilibria Relevant to the Deep Earth
Phase diagrams illustrate the stability of phases in Pressure,Temperature, chemical activity space. Many igneous and metamorphic systems contain up to 11 significant chemical components. This means that a full depiction of phase equilibria in these systems requires 13 dimension. Petrologists have devised a host of sections, pseudo sections and projections to create illustrative 2D diagrams from these multi-dimensional spaces. Mineral physics has mostly confined itself to working in relatively simple chemical systems (e.g. MgO-SiO2) that serve as a proxy for the chemically more complex Earth. Another strategy is to look at changes with pressure and temperature in a single bulk composition (e.g. MORB or pyrolite). A selection of relevant phase diagrams is given below. A note regarding the names of phases
For conversational convenience, some mineralogists and nearly all mineral physicists use the names of common minerals to refer to other compounds with the same structure. So for example, we use 'spinel' to refer to A2BO4 phases that are isostructural with the mineral spinel (MgAl2O4) and 'perovskite' to refer to phases that are isostructural with the mineral perovksite (CaTiO3). Other conventions for naming phases come out of material science and solid state physics. So one commonly sees Greek letters used to refer to polymorphs of a given compound (e.g. α, β, γ, δ, ε). Phases may also be referred to by their packing arrangement; for example BCC for body centered cubic structure, FCC for face centered cubic and HCP for hexagonal close packed. Most high pressure phases were discovered in the lab and bear these sorts of names. However, some have subsequently been identified in meteorites and have been given proper mineral names like Akimotoite and Ringwoodite. This has led to some phases having multiple names (e.g. Ringwoodite = γ-olivine = spinel); which may be somewhat confusing for the uninitiated.(Mg,Fe)2SiO4
(Mg,Fe)2SiO4 is a major constituent of the rocky portion of the planet. At low pressure it occurs as the familiar mineral olivine. Most mineralogy texts contain good descriptions of the physical and chemical properties of olivine. The "Orthosilicates" volume of Reviews in Mineralogy (volume 5) provides a comprehensive review. With increases in pressure the olivine structure become unstable relative to Wadsleyite and Ringwoodite which are denser. The transitions in the earth occur at ~410 km and ~520 km. Since this is a two component system, each transition is accompanied by a two phase loop. At ~670 km depth, Ringwoodite disproportionates into a perovskite structured phase (usually just called perovskite) and magnesiowustite (which is sometimes referred to as periclase (the name of the Mg end-member) or in some cases rocksalt (which is the name of the structure)). To simplify life researchers may choose to work in the pure endmember Mg2SiO4, (forsterite olivine) or Fe2SiO4 (fayalite olivine). A particularly popular starting material for (Mg,Fe)2SiO4 is gem quality peridote from San Carlos, AZ which is referred to as San Carlos olivine.


(Mg,Fe)SiO3
(Mg,Fe)SiO3 is another major constituent of the mantle. At low pressure it occurs as an orthopyroxene, it converts to clinopyroxene with increasing pressure. Like olivine there are many good resources describing the physics and chemical properties of pyroxenes, the "Pyroxenes" volume of Reviews in Mineralogy (volume 7) is a good place to start. With pressure pyroxenes convert to majorite garnet, or disproportionates to or wadsleyite + stishovite depending on the temperature. These assemblages convert to either the perovskite or illmenite structure (depending on P,T conditions) and finally a relatively newly discovered compound referred to as the post-perovskite structure.
The pyroxene structure can accept a wider range of cations including large cations such at Na, therefore pyroxene is thought to host much of the chemical diversity in the shallow upper mantle. Once it forms, majorite garnet takes on this role. The composition of the mantle is thought to lie somewhere between olivine (Mg2SiO4) and pyroxene (MgSiO3) on the MgO-SiO2 join. The ratio of olivine to pyroxene is still up for debate, but pyroxene is either the most abundant phase in the shallow upper mantle or the second most abundant. Again there are a plethora of names for the phases in this system. Enstatite is the mineral name for low Ca Mg orthopyroxene. The abbreviation 'OPX' and 'CPX' are often used for ortho- and clinopyroxene. Majorite is usually referred to simply as 'garnet'. Another thing to be aware of is that at low pressure there is an miscibility gap between Fe- and Mg- orthopyroxene and Ca-bearing clinopyroxene (usually referred to by the name of its Mg,Ca- endmember, diopside). So when 'clinopyroxene' is used one must figure out whether the low pressure Ca-bearing variety or the high pressure pyroxene is being referred to.


SiO2
The low pressure polymorphs of quartz (low and high quartz, cristobalite and tridymite) are familiar to anyone with a geology background since they are common crustal minerals. Coesite occurs less commonly in crustal rocks but is of great interest when it occurs, as it has interesting tectonic implications. Stishovite has the same structure as rutile (TiO2) and has a mineral name because it is found in meteorite impact sites. At very high pressure stishovite transforms to a CsCl structure and then to one of the structures exhibited by PbO2; the orthorhombic α-PbO2 structure. The relative importance of SiO2 in the deep earth depends on the composition of the lower mantle, which like much things about the deep earth, is still up for debate.



Hydrous Assemblages
The role and mineralogical home of water in the mantle is a topic of great interest and debate. Significant amounts of water may be contained in anhydrous phases but the presence of water may also stabilize a complex series of hydrous phases. The key features to be aware of in mantle phase equilibria dealing with water are 1) the effect of water on the solidous 2) the change from increasing to decreasing temperature stability of hydrous phases with pressure at low to moderate pressure and 3) increasing thermal stability of hydrous phases with pressure above the so called "choke point" at ~5 GPa.

Realistic Mantle Compositions
The composition of the mantle has been modeled as close to the composition of peridotite, as pyrolite (an enriched source which produced basalt and peridotite), as piclogite (with a more Si and Fe rich lower mantle), and as chondrite (the composition of stony meteorites that have not been modified by melting or differentiation). In all cases at least 5 or 6 oxide components are required to describe the system. This vastly complicates phase diagrams because they can no longer be depicted with 2D diagrams without making projections of some sort. Fortunately the broad outline of phase relations in realistic mantle compositions are captured by the phase diagrams for Mg2SiO4 and MgSiO3.
Fe
The core is thought to consist of an Fe-Ni alloy with some mixture of other light elements including S. Phase relations in the pure Fe system are often used as a proxy for phase relations in the core.
Resources for Learning About Phase Equilibria
SERC has an extensive website on Teaching Phase Equilibria which has many tutorial pages suitable for students as well as instructors.
Gibbs Phase Rule: Where it all Begins
Modeling programs (MELTS, TWQ, Perplex, THERMOCALC and Theriak-Domino)
Method of Schreinemakers -- A Geometric Approach to Constructing Phase Diagrams
Other useful sites include:
Thermodynmics and Kinetics from MIT OPEN courseware (readings, lecture notes and videos of lectures) as well as
Tim Grove's Thermodynamics for Geoscientists course which has a focus on the phase equilibria of melts.
References Cited and Additional Literature
Bolfan-Casanova, N., Andrault, D., Amiguet, E. and Guignot, N., 2009. Equation of state and post-stishovite transformation of Al-bearing silica up to 100 GPa and 3000K, Physics of the Earth and Planetary Interiors, 174, 70-77.
Burnley, P. C. and Navrotsky, A., 1996, Synthesis of high-pressure hydrous magnesium silicates: observations and analysis. American Mineralogist. v 81, pp. 317-326.
Fei, Y., and C. M. Bertka, Phase transitions in the Earth's mantle and mantle mineralogy, in Mantle Petrology: Field Observations and High Pressure Experimentation, Y. Fei, C. M. Bertka, and B. O. Mysen, eds., pp. 189-207, Special Publication No. 6, Geochemical Society, Houston, 1999.
Komabayashi, T., and Fei, Y., 2010. Internally consistent thermodynamic database for iron to the Earth's core conditions, Journal of Geophysical Research, 115.
Kung, J., Li, B., Uchida, T., Wang, Y., Neuville, D., and Liebermann, R., 2004. Insitu measurements of sound velocities and densities across the orthopyroxene high-pressure clinopyroxene transition in MgSiO3 at high pressure. Physics of the Earth and Planetary Interiors, 147, 27–44.
Litasov, K., Ohtani, E., Sano, A., Suzuki, A. and Funakoshi, K., 2005. In situ X-ray diffraction study of post-spinel tranformation in a peridotite mantle: Implication for the 660-km discontinuity, Earth and Planetary Science Letters, 238, 311-328.
Liu, L., and Bassett, W.A., Elements, Oxides, and Silicates: High-pressure Phases with Implications for the Earth's Interior. New York: Oxford University Press, 1986.
Murakami, M., and Bass, J.D., 2011. Evidence of denser MgSiO3 glass above 133 gigapascal (GPa) and implications for remnants of ultradense silicate melt from a deep magma ocean, Proc. Nat. Acad. Sci., 108 (42) 17286-17289.
Murakami, M., Hirose, K., Ono, S., and Ohishi, Y., 2003. Stability of CaCl2-type and α-PbO2-type SiO2 at high pressure and temperature determined by in-situ X-Ray measurements, Geophysical Research Letters, 30(5).
Murakami, M., Hirose, K., Sata, N., and Ohishi, Y., 2005. Post-perovskite phase transition and mineral chemistry in the pyrolitic lowermost mantle, Geophysical Research Letters, 32.
Navrotsky, A., 1989. Silicates and Germanates at High Pressure, Solid State Ionics, 32-33, 288-297.
Ohtani, E., Recent Progress in Experimental mineral Physics: Phase Relations of Hydrous Systems and the Role of Water in Slab Dynamics, in Earth's Deep Mantle: Structure, Composition, and Evolution. Geophysical Monograph Series 160, R.D. van der Hilst, J.D. Bass, J. Matas, and J. Trampert, eds., American Geophysical Union, p 321-334, 2006.
Ulmer, P., and Trommsdorff, V., 1995. Serintine Stability to Mantle Depths and Subduction-Related Magmatism, Science, 268(5212), PP. 858-861.
Acknowledgements
These materials are being developed with the support of COMPRES, the Consortium for Materials Properties Research in Earth Sciences, under NSF Cooperative Agreement EAR 10-43050 and is partially supported by UNLV's High Pressure Science and Engineering Center, a DOE NNSA Center of Excellence supported under DOE NNSA Cooperative Agreement No. DE FC52-06NA26274.