
X-ray Powder Diffraction (XRD)
This resource was originally developed for the Integrating Research and Education (IntRE) project, and the original resource can be found on the IntRE XRD page.
Barbara L Dutrow, Louisiana State University, Christine M. Clark, Eastern Michigan University
What is X-ray Powder Diffraction (XRD)
X-ray powder diffraction (XRD) is a rapid analytical technique primarily used for phase identification of a crystalline material and can provide information on unit cell dimensions. The analyzed material is finely ground, homogenized, and average bulk composition is determined.
Fundamental Principles of X-ray Powder Diffraction (XRD)
Max von Laue, in 1912, discovered that crystalline substances act as three-dimensional diffraction gratings for X-ray wavelengths similar to the spacing of planes in a crystal lattice. X-ray diffraction is now a common technique for the study of crystal structures and atomic spacing.
X-ray diffraction is based on constructive interference of monochromatic X-rays and a crystalline sample. These X-rays are generated by a cathode ray tube, filtered to produce monochromatic radiation, collimated to concentrate, and directed toward the sample. The interaction of the incident rays with the sample produces constructive interference (and a diffracted ray) when conditions satisfy Bragg's Law (nλ=2d sin θ). This law relates the wavelength of electromagnetic radiation to the diffraction angle and the lattice spacing in a crystalline sample. These diffracted X-rays are then detected, processed and counted. By scanning the sample through a range of 2θangles, all possible diffraction directions of the lattice should be attained due to the random orientation of the powdered material. Conversion of the diffraction peaks to d-spacings allows identification of the mineral because each mineral has a set of unique d-spacings. Typically, this is achieved by comparison of d-spacings with standard reference patterns.
All diffraction methods are based on generation of X-rays in an X-ray tube. These X-rays are directed at the sample, and the diffracted rays are collected. A key component of all diffraction is the angle between the incident and diffracted rays. Powder and single crystal diffraction vary in instrumentation beyond this.
X-ray Powder Diffraction (XRD) Instrumentation - How Does It Work?
X-ray diffractometers consist of three basic elements: an X-ray tube, a sample holder, and an X-ray detector.

The geometry of an X-ray diffractometer is such that the sample rotates in the path of the collimated X-ray beam at an angle θ while the X-ray detector is mounted on an arm to collect the diffracted X-rays and rotates at an angle of 2θ. The instrument used to maintain the angle and rotate the sample is termed a goniometer. For typical powder patterns, data is collected at 2θ from ~5° to 70°, angles that are preset in the X-ray scan.
Applications
X-ray powder diffraction is most widely used for the identification of unknown crystalline materials (e.g. minerals, inorganic compounds). Determination of unknown solids is critical to studies in geology, environmental science, material science, engineering and biology.
Other applications include:
- characterization of crystalline materials
- identification of fine-grained minerals such as clays and mixed layer clays that are difficult to determine optically
- determination of unit cell dimensions
- measurement of sample purity
With specialized techniques, XRD can be used to:
- determine crystal structures using Rietveld refinement
- determine of modal amounts of minerals (quantitative analysis)
- characterize thin films samples by:
- determining lattice mismatch between film and substrate and to inferring stress and strain
- determining dislocation density and quality of the film by rocking curve measurements
- measuring superlattices in multilayered epitaxial structures
- determining the thickness, roughness and density of the film using glancing incidence X-ray reflectivity measurements
- make textural measurements, such as the orientation of grains, in a polycrystalline sample
Strengths and Limitations of X-ray Powder Diffraction (XRD)?
Strengths
- Powerful and rapid (< 20 min) technique for identification of an unknown mineral
- In most cases, it provides an unambiguous mineral determination
- Minimal sample preparation is required
- XRD units are widely available
- Data interpretation is relatively straight forward
Limitations
- Homogeneous and single phase material is best for identification of an unknown
- Must have access to a standard reference file of inorganic compounds (d-spacings, hkls)
- Requires tenths of a gram of material which must be ground into a powder
- For mixed materials, detection limit is ~ 2% of sample
- For unit cell determinations, indexing of patterns for non-isometric crystal systems is complicated
- Peak overlay may occur and worsens for high angle 'reflections'
User's Guide - Sample Collection and Preparation
Determination of an unknown requires: the material, an instrument for grinding, and a sample holder.
- Obtain a few tenths of a gram (or more) of the material, as pure as possible
- Grind the sample to a fine powder, typically in a fluid to minimize inducing extra strain (surface energy) that can offset peak positions, and to randomize orientation. Powder less than ~10 μm(or 200-mesh) in size is preferred
- Place into a sample holder or onto the sample surface:
 Packing of fine powder into a sample holder. Details
Packing of fine powder into a sample holder. Details- smear uniformly onto a glass slide, assuring a flat upper surface
- pack into a sample container
- sprinkle on double sticky tape
- Care must be taken to create a flat upper surface and to achieve a random distribution of lattice orientations unless creating an oriented smear.
- For analysis of clays which require a single orientation, specialized techniques for preparation of clay samples are given by USGS.
- For unit cell determinations, a small amount of a standard with known peak positions (that do not interfere with the sample) can be added and used to correct peak positions.

Data Collection, Results and Presentation
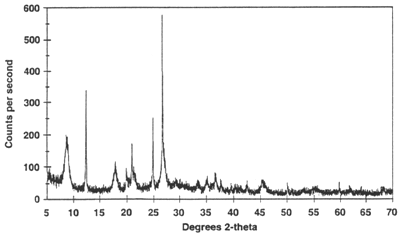
Data Collection The intensity of diffracted X-rays is continuously recorded as the sample and detector rotate through their respective angles. A peak in intensity occurs when the mineral contains lattice planes with d-spacings appropriate to diffract X-rays at that value of θ. Although each peak consists of two separate reflections (Kα1 and Kα2), at small values of 2θ the peak locations overlap with Kα2 appearing as a hump on the side of Kα1. Greater separation occurs at higher values of θ. Typically these combined peaks are treated as one. The 2λ position of the diffraction peak is typically measured as the center of the peak at 80% peak height.
Data Reduction
Results are commonly presented as peak positions at 2θ and X-ray counts (intensity) in the form of a table or an x-y plot (shown above). Intensity (I) is either reported as peak height intensity, that intensity above background, or as integrated intensity, the area under the peak. The relative intensity is recorded as the ratio of the peak intensity to that of the most intense peak (relative intensity = I/I1 x 100 ).
Determination of an Unknown
The d-spacing of each peak is then obtained by solution of the Bragg equation for the appropriate value of λ. Once all d-spacings have been determined, automated search/match routines compare the ds of the unknown to those of known materials. Because each mineral has a unique set of d-spacings, matching these d-spacings provides an identification of the unknown sample. A systematic procedure is used by ordering the d-spacings in terms of their intensity beginning with the most intense peak. Files of d-spacings for hundreds of thousands of inorganic compounds are available from the International Centre for Diffraction Data as the Powder Diffraction File (PDF). Many other sites contain d-spacings of minerals such as the American Mineralogist Crystal Structure Database. Commonly this information is an integral portion of the software that comes with the instrumentation.
Determination of Unit Cell Dimensions
For determination of unit cell parameters, each reflection must be indexed to a specific hkl.
Literature
The following literature can be used to further explore X-ray Powder Diffraction (XRD)
- Bish, DL and Post, JE, editors. 1989. Modern Powder Diffraction. Reviews in Mienralogy, v. 20. Mineralogical Society of America.
- Cullity, B. D. 1978. Elements of X-ray diffraction. 2nd ed. Addison-Wesley, Reading, Mass.
- Klug, H. P., and L. E. Alexander. 1974. X-ray diffraction procedures for polycrystalline and amorphous materials. 2nd ed. Wiley, New York.
- Moore, D. M. and R. C. Reynolds, Jr. 1997. X-Ray diffraction and the identification and analysis of clay minerals. 2nd Ed. Oxford University Press, New York.
Related Links
For more information about X-ray Powder Diffraction (XRD) follow the links below.
- For more information on XRD Methods, visit the USGS website
- For additional information on X-ray basics (more info) ; Materials Research Lab, University of California- Santa Barbara
- Rigaku Journal; an on-line journal that describes and demonstrates a wide range of applications using Xray diffraction.
- Cambridge University X-ray Tutorial
- International Union of Crystallography (IUCr) Teaching Pamphlets
- Introduction to X-ray Diffraction (more info) --University of California, Santa Barbara
- Introduction to Crystallography --from LLNL
- X-ray Crystallography Lecture Notes ( This site may be offline. ) --from Steve Nelson, Tulane University
- Reciprocal Net--part of the National Science Digital Library. Use the "Learn About" link to find animations of the structures of common molecules (including minerals), crystallography learning resources (tutorials, databases and software), resources on crystallization, and tutorials on symmetry and point groups.
Teaching Activities and Resources
Teaching activities, labs, and resources pertaining to X-ray Powder Diffraction (XRD).
- X-ray techniques lab exercises from the SERC Teaching Mineralogy Collections
- Weathering of Igneous, Metamorphic, and Sedimentary Rocks in a Semi-Arid Climate - An Engineering Application of Petrology - This problem develops skills in X-ray diffraction analysis as applied to clay mineralogy, reinforces lecture material on the geochemistry of weathering, and demonstrates the role of petrologic characterization in site engineering.
- Teaching Guide to X-ray Diffraction at Cambridge
- A Powerpoint presentation on use of XRD in Soil Science (PowerPoint 1.6MB Sep7 07) by Melody Bergeron, Image and Chemical Analysis Laboratory at Montana State University.
- Brady, John B., and Boardman, Shelby J., 1995, Introducing Mineralogy Students to X-ray Diffraction Through Optical Diffraction Experiments Using Lasers. Jour. Geol. Education, v. 43 #5, 471-476.
- Brady, John B., Newton, Robert M., and Boardman, Shelby J., 1995, New Uses for Powder X-ray Diffraction Experiments in the Undergraduate Curriculum. Jour. Geol. Education, v. 43 #5, 466-470.
- Dutrow, Barb, 1997, Better Living Through Minerals X-ray Diffraction of Household Products, in: Brady, J., Mogk, D., and Perkins D. (eds.) Teaching Mineralogy, Mineralogical Society of America, p. 349-359.
- Hovis, Guy, L., 1997, Determination of Chemical Composition, State of Order, Molar Volume, and Density of a Monoclinic Alkali Feldspar Using X-ray Diffraction, in: Brady, J., Mogk, D., and Perkins D. (eds.) Teaching Mineralogy, Mineralogical Society of America, p. 107-118.
- Brady, John B., 1997, Making Solid Solutions with Alkali Halides (and Breaking Them) , in: Brady, J., Mogk, D., and Perkins D. (eds.) Teaching Mineralogy, Mineralogical Society of America, p. 91-95.
- Perkins, Dexter, III, and Sorensen, Paul, Mineral Synthesis and X-ray Diffraction Experiments, in: Brady, J., Mogk, D., and Perkins D. (eds.) Teaching Mineralogy, Mineralogical Society of America, p. 81-90.
- Hollecher, Kurt, A Long-Term Mineralogy Practical Exam, in: Brady, J., Mogk, D., and Perkins D. (eds.) Teaching Mineralogy, Mineralogical Society of America, p. 43-46.
- Moecher, David, 2004, Characterization and Identification of Mineral Unknowns: A Mineralogy Term Project, Jour. Geoscience Education, v 52 #1, p. 5-9.
- Hluchy, M.M., 1999, The Value of Teaching X-ray Techniques and Clay Mineralogy to Undergraduates, Jour. Geoscience Education, v. 47, p. 236-240.